Download
Please download the following single file for the software. (Total size: 284KB)
Linux/Unix version: MSS (mss.tar.gz)
The package contains all the required programs and scripts. Currently, it only has the Linux/Unix version. Installation of the software requires the stardard development tools "make" and "g++". If you want to have graphical outputs, you should have gnuplot installed.
Installation
To install the software, download the file mss.tar.gz and follow the steps below.
- Unzip the file by gunzip mss.tar.gz. You will get mss.tar.
- Extract the file by tar -xzvf mss.tar. You will get 3 directories (data, sample and source) and two files (Makefile and mss.sh)
- Compile the programs by make.
Input File Formats
The input genomes should be stored in files in FASTA format.
Sample Input Sequence Files: Bm.fasta, Xc.fasta
Normal Usage of the Software
For normal users, the default values of the software should be fine for most of the cases (please see the section at the bottom for advanced usage of the software). For the normal usage, the only parameters needed for the program are:- The two input file names.
- The flag for aligning sequences in DNA level or in translated protein level.
- To align in DNA level, please add the '-D' flag.
./mss.sh -D Bm.fasta Xc.fasta - To align in translated protein level, no flag is needed.
./mss.sh Bm.fasta Xc.fasta
Output Formats
The software produces outputs in two formats. One is a graphical display of the potential gene regions (in ps format), and the other is in MUM file format.
-
Graphical Display of Potential Gene Regions
One postscript file will be generated by the default setting of the software.
Sample Output ps File: BmXc.mum2.ps
Each red line represents a MUM pair with same orientation in both genomes, while each green line represents a MUM pair with different orientations.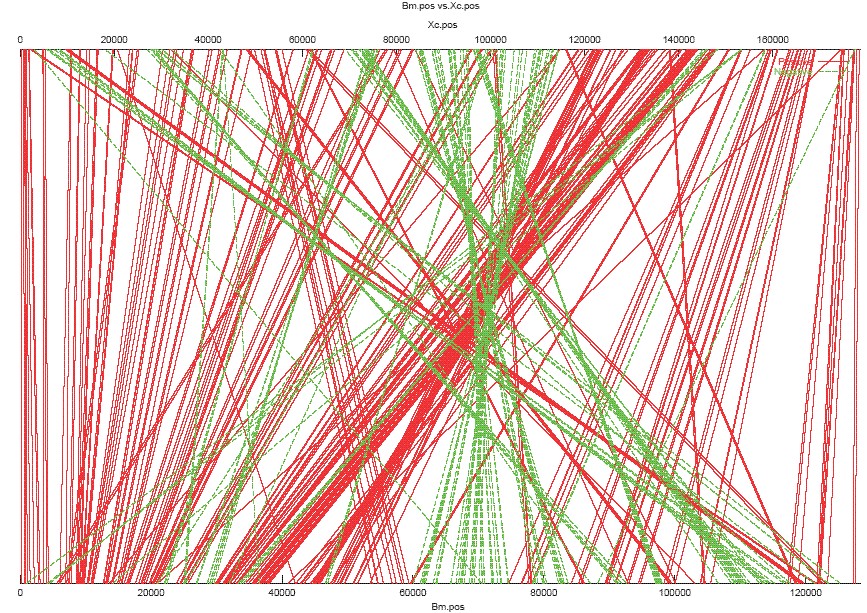
- MUMs (or clustered MUMs)
The MUM file is primarily a 6-column list. Each line in the file represents a MUM and each of the six columns correponds to an attribute of that MUM. Lines are in the format of weight s1 s2 e1 e2 sign, where
- weight is the weight of the MUM. Note that the weight may not equal to the length of the MUM.
- s1 and e1 are the start and end positions of the MUM in sequence 1.
- s2 and e2 are the start and end positions of the MUM in sequence 2.
- sign is the sign of the MUM.
Sample Output MUM File: out.h16.2
Advanced Usage of the Software
Now we will explain the meanings of all the different parameters here, since there are quite a number of
parameters needed for the software. We expect only advanced users will use these options, since default settings work
fine in most cases.
- Path of the input files
Note: Currently our software does not support wildcards or pathname expansion. So typing seq*.txt will not map to seq1.txt or seq2.txt. Instead, a File Not Found error will be reported. - Parameters of the programs, including
- General Options:
1. -o pfx Set the prefix of the output files (default: "out") 2. -D Align genomes in DNA level (default: align in protein level) 3. -l num Set the minimum length of an exact match for the 1st filtering process (default: 9) 4. -f num Set the gap distance for the 1st filtering process (default: 6000) 5. -L num Set the minimum length of an exact match for the 2nd filtering process (default: 12) 6. -F num Set the gap distance for the 2nd filtering process (default: 6000) 7. -w num Set the weight-factor for the output of MaxMinCluster (default: 1.4) 8. -U Do not generate graphical output (default: graphical outputs are genereated) 9. -h Show all possible options - MaxMinCluster Options:
1. -M Use the gap model as the distance requirement. (default: the fraction model is used) 2. -k num Set the noise level of the clustering algorithm (default: 3) 3. -g num Set the maximum gap of matches between a cluster (default: 2000) 4. -s num Set the minimum size of a cluster (default: 8) 5. -d num Set the gap difference for the fraction model (default: 5) 6. -r num Set the gap_ratio for the fraction model (default 0.1) - MSS Options:
1. -t num Set the gap_threshold of matches between a cluster (default: 2.7) 2. -m num Set the maximum number of mutations allowed (default: 40)
- General Options: